The Molecular Genomics Of Metastatic Brain Tumors
Studying the molecular genomics of metastatic brain tumors aids in identifying patients at risk of developing one. Improved understanding is needed to avoid dismal patient outcomes.
Author:Suleman ShahReviewer:Han JuJan 25, 202413 Shares6.6K Views
Studying the molecular genomics of metastatic brain tumorscan be a complex task.
Metastatic brain tumors remain an intractable clinical problem despite notable advances in the treatment of the primary cancers.
It is estimated that 30 to 40% of breast and lung cancer patients will develop brain metastases.
Typically, brain lesions are not diagnosed until patients exhibit neurological symptoms because there are currently no tests that can predict which patients will be afflicted.
Brain metastases are resistant to current chemotherapies, and despite surgical resection and radiotherapy, the prognosis for these patients remains very poor with an average survival of only 6-9 months.
Cancer is ultimately a genetic disease, involving patient genetics and aberrant tumor genomics; therefore, the pursuit of an explanation for why or how brain metastases occur requires investigation of the associated somatic mutations.
Incidence And Current Therapeutic Options
There are approximately 200,000 newly diagnosed cases of brain metastases annually in the United States, 10-fold greater than primary brain cancer.
Current estimates indicate that up to 40% of patients with a systemic primary cancer will develop a metastatic brain tumor (MBT).
A majority of brain metastases come from:
- lung cancer (50-60%)
- breast cancer (20-30%)
- melanoma (5-10%)
- various cancers (including gastrointestinal, esophageal, prostate, and ovarian), with combined contribute of 5-10%
Improved treatments for primary cancers are allowing patients to live longer, leading to increased incidence of MBTs.
Brain metastases are among the most feared complications for cancer patients because of diminished quality of lifeand a lack of effective chemotherapies.
There are no predictive biomarkers to identify patients at risk to develop brain metastasis, and the healthcare system cannot bear the burden to screen all cancer patients for early detection of MBTs.
Metastatic lesions are discovered when a patient presents with neurological symptoms; then, if left untreated, median survival is 1 month.
The standard course of treatment for patients with controlled systemic disease is microsurgical resection plus radiation (whole-brain radiation therapyand/or stereotactic radiosurgery), extending median survival to 9-10 months.
Patients receiving radiation alone have a median survival of 4-6 months.
Chemotherapeutic agents are not effective, partly attributed to their limited ability to penetrate the blood-brain barrier (BBB).
Although new therapies are in development to penetrate the BBB, recent evidence also suggests that the BBB may not be intact in patients with brain tumors.
Promising preliminary studies indicate that molecularly targeted therapies (small-molecule kinase inhibitors against activated oncogenes) can cross the BBB and correlate with modestly improved outcomes.
Selected independent case studies report marked tumor regression and sustained response.
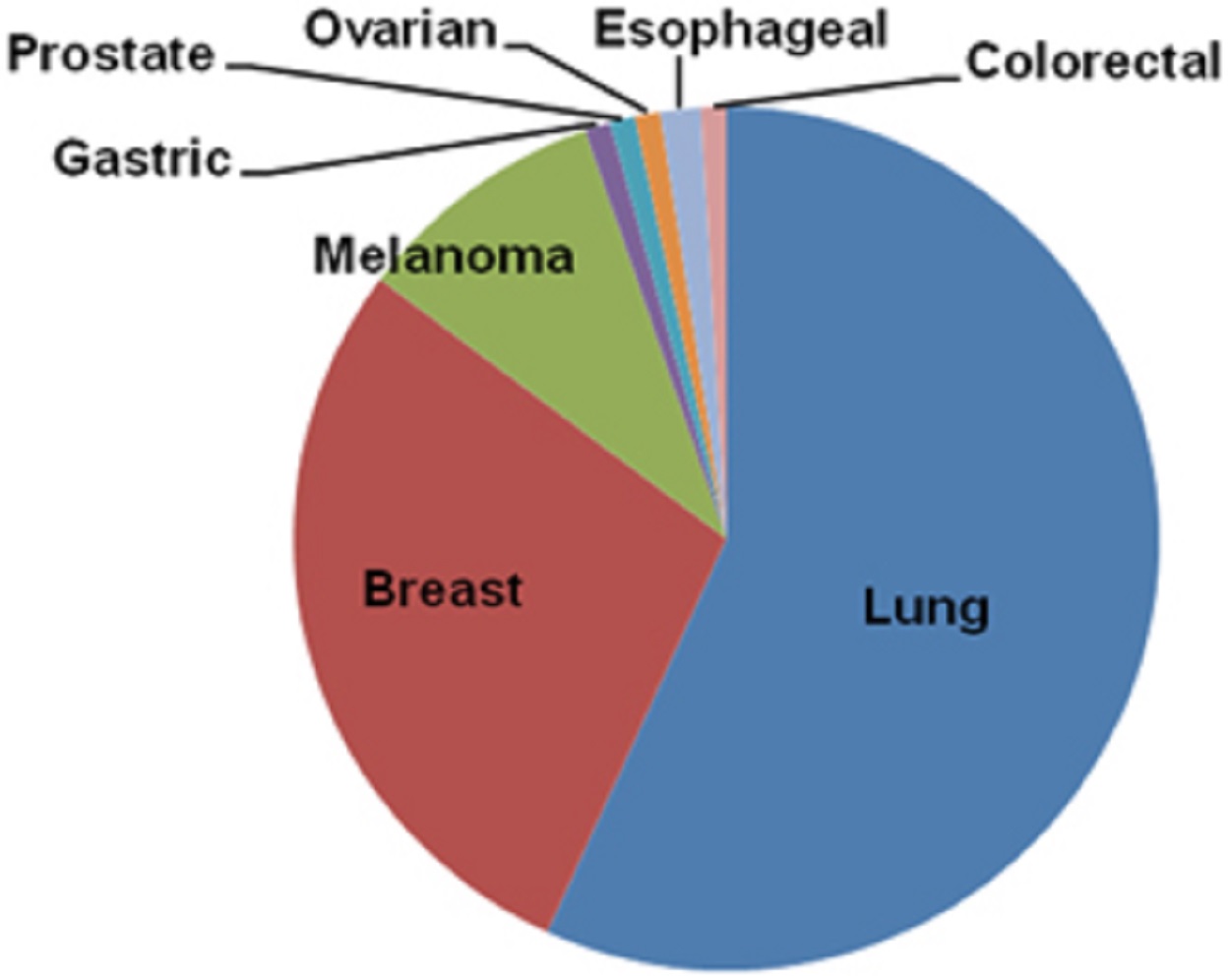
The vast majority of MBTs develop from primary lung and breast cancers.
The frequency at which lung and breast cancer patients develop brain metastases is approximately 25% and approximately 30%, respectively, but differs more specifically according to the histology or molecular subtypes of the systemic disease.
In the study published in the journal Cancer and Metastasis Reviewsin 1989, the author, Stephen Paget, first posited that the nonrandom pattern of metastasis linked to specific primary cancers was not due to chance and that formation of metastatic tumors depends on interactions between the microenvironment of the metastatic site and the metastatic cancer cells.
Herein, we focus our review on this broadly accepted concept and what is known about the stages of metastasis as they relate to the understanding that cancer is ultimately a genetic disease with pathological phenotypes driven by oncogenic, somatic mutations.
Stages In The Metastatic Process
Metastatic biological processes, including the role of microenvironment and the ability of cancer cells to transverse the BBB, have been extensively reviewed in great detail elsewhere.
Briefly, the stages of metastasis comprise, in temporal order of occurrence:
- local invasion
- survival in the circulation
- intravasation
- extravasation
- micrometastasis formation
- metastatic colonization
Invasion depends upon an epithelial to mesenchymal transition (EMT) where epithelial cell traits are suppressed and mesenchyme cell traits are activated, losing cell polarity and cell-cell adhesion.
Underlying EMT is a gene expression program regulated by epigenetic changes across the genome and aberrant expression of key transcription factors and microRNAs.
New evidence describes further complexity, where a reversal of the EMT process is required for colonized metastatic cells to proliferate.
This potential phenotypic plasticity is consistent with epigenetic principles inherent to the hierarchically organized cancer stem cell model, where cancer stem cellsundergo irreversible epigenetic changes to produce phenotypically diverse cancer cells.
Additional work does suggest that with the EMT process, cancer cells also acquire stem cell-like traits.
The resulting “cancer stem cells” represent a minor subset of cells within a tumor.
They maintain the capability for unlimited self-renewal, have the ability to seed new tumors and are a potential cell type from the primary tumor that initiates a metastatic lesion.
Relatively large numbers of circulating tumor cells can be detected in the blood of carcinoma patients, including those who do not clinically develop distant metastases.
Of those numbers of circulating cells, approximately 80% undergo extravasation.
However, once implanted at distant sites, fewer than 3% of cancer cells survive to form micrometastases and fewer than 0.1% persist to metastatic macro-colonization.
Macro-colonization is described as the rate-limiting step in metastatic tumor formation.
A related mechanistic hypothesis, consistent with the clinical observation of long latency periods for distant relapse following initial diagnosis, posits that during the latent period, cancer cells, and likely also the surrounding microenvironment, undergo gradual evolution to acquire molecular aberrations advantageous for metastatic colonization either because micrometastatic cell growth and death are equal or by discontinuous growth and periods of quiescence.
However, for brain metastasis from lung and breast cancers, the latency period is relatively short.
The median latency from breast cancer diagnosis to MBTs is 34 months (with HER2 positive developing more quickly) and lung cancer-derived MBTs are detected within months of the initial diagnosis.
This suggests a different course for MBTs in which only a subset of cells (possibly stem cells) in the primary tumor has the molecular prerequisites for all metastatic stages.
Alternative to a stem cell hypothesis, selection pressures within progressing primary tumors give rise to clonal evolution of tumor cells with acquired genetic variations and increased malignant or metastatic potential.
In line with this hypothesis, metastatic potential and inherent resistance against selective pressures are coincident with heightened genetic instability.

The unique environment of the brain compared to other organs presents an optimum environment for metastatic progenitor cells arising from particular tissues, fitting with the nonrandom pattern of metastasis.
While tropism and anatomical limitations are also thought to be pre-determinants, strong evidence suggests that the dominant influence is the nature of compatibility between the primary cell of origin and the site of metastatic colonization.
In a study published in the journal Cancer Researchin 1980, authors Ian R. Hart and Isaiah J. Fidler established this with a rodent xenograft model of human metastatic melanoma in which the same number of micrometastases were identified in the parenchyma of lung and kidney tissues embedded side by side, but metastatic tumors only developed in the lung fragment and not in the kidney fragment.
Details of the biology of brain metastases are still emerging, but it appears that multiple widely expressed mechanisms in cancer cells are able to support intravasation, survival in circulation, extravasation and micrometastasis.
However, a more select oncogenic mechanism drives colonization and growth.
Putative oncogenic drivers of brain metastasis and whether they exist in the primary tumor or are acquired in disseminated cells are further addressed below.
Oncogenes And Tumor Suppressors
Decades of research have contributed to the understanding that carcinogenesis is governed by the activation of oncogenes and inactivation of tumor suppressor genes.
Oncogenes are activated by various mechanisms including gain-of-function sequence mutations, overexpression due to genomic amplification or deregulated epigenetics and genomic rearrangements.
Tumor suppressors, factors that otherwise slow normal cell growth processes or promote tissue differentiation, are inactivated by genomic lesions resulting in loss-of-function.
Thus, within the tumor genome, the causes for initiation and progression of the disease can be found.
Important clinical advances in primary cancer treatments are attributed to molecularly targeted treatments tailored to target activated oncogenes, for example:
| BRAF | HER2 |
| EGFR | MET |
Their aberrant presentation in the primary tumor genome presents a biomarker to indicate the efficacy of the targeted treatment.
Moreover, oncogenic factors often possess kinase or ligand-binding functions that present “druggable” protein motifs.
The benefits of targeted treatments over traditional chemotherapy are clear, resulting in:
- greater efficacy
- less debilitating side effects
The success of driver oncogene-targeted therapies against specific primary cancers can also be attributed to the fact that oncogene-directed signaling promotes the many phenotypes that define the pathobiology of cancer cells, including:
- unrestricted growth
- immune system evasion
- metabolic transformation
- invasive potential
- cancer cell tumorigenicity (local and metastatic)
This wide array of biological impacts suggests that somatic genome mutations affecting activated oncogenes and inactivated tumor suppressors are likely causally implicated in all stages of MBT development.
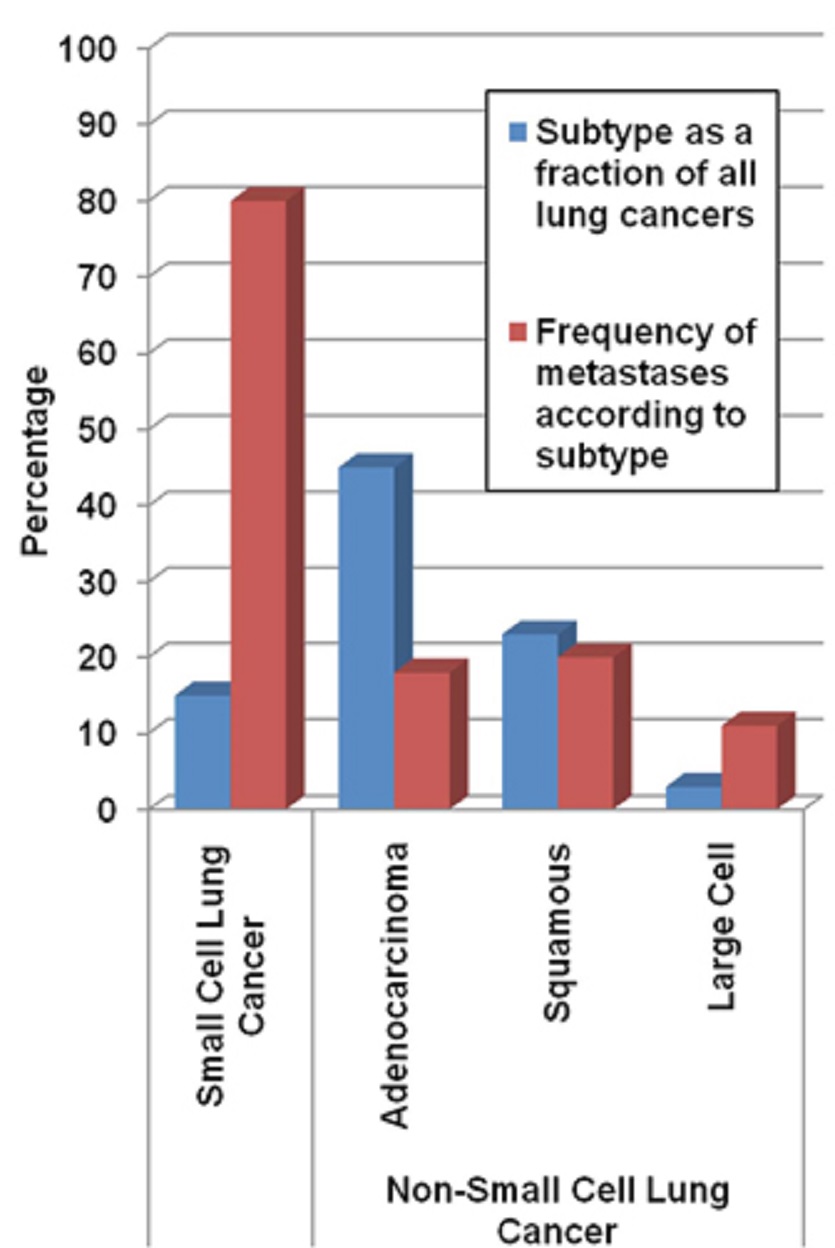
Genomic Mutation Landscape Of Metastatic Brain Tumors
Early studies indicate that molecularly targeted therapies in the form of small-molecule kinase inhibitors and monoclonal antibodies against activated oncogenes can elicit a response in the treatment of metastatic tumors and potentially reduce the rates of metastatic recurrence.
On the other hand, increased incidence of MBTs in patients with controlled primary cancer, and peripheral metastasis has also been attributed to the inefficiency of targeted drug transfer across the BBB.
Although further investigation is required, each of these data indicates that an oncogene mechanistically linked to the primary cancer is also implicated in driving the incidence of MBTs.
This scenario may apply to the mutated expression of BRAF oncogene in:
- metastases from melanoma
- aberrant levels of HER2 oncogene in breast cancer brain metastases
- mutated EGFR oncogene in lung cancer brain metastases
The potential importance of BRAF, HER2, and EGFR for brain metastasis was identified based on knowledge of their driver oncogene status and frequency of mutation in the related primary cancer.
Their specific oncogenic role and frequency of mutation in MBTs remain to be ascertained.
Additionally, it is unclear if for a given oncogene certain types of activating mutations are more often represented in brain metastases, as early evidence suggests.
Studying these unknowns will explain why brain metastases develop in cases where systemic disease is treated with targeted therapies and why treating MBTs has had only modest effects and no impact on therapy options.
An important starting point is comprehensive genomic characterization of MBTs compared to their primary cancers, particularly the well-characterized molecular breast and lung cancers subtypes.
Despite the large number of patients afflicted, the characterization of significant cancer-linked genomic copy number aberrations, rearrangements, and sequence mutations in MBTs is only in its infancy.
Gene Coding Mutations
Most studies to date that have examined somatic DNA coding mutations in MBTs have been biased towards the activating mutations in oncogenes or tumor suppressors previously associated with the primary cancer.
A limited study that included DNA from 10 melanoma metastases measured a panel of BRAF, NRAS, AKT, PIK3CA ,and KIT activating mutations using a highly sensitive primer extension.
Mass spectrometry approach showed that BRAF activating mutations were the most frequent and secondarily NRAS, similar to primary melanoma.
A later study using Sanger sequencing identified BRAF and NRAS mutations in both primary melanoma and matched brain metastases from 44 patients at 80% consistency.
Another study measured mutations in 19 oncogenes, including BRAF, KRAS, NRAS, and PIK3CA, by high-resolution DNA melting analysis of specimens from matched brain metastases and primary colorectal cancer.
The study recapitulated that KRAS mutations were a more common event in colorectal cancer and that 9 of 10 matched brain metastases were 100% concordant with mutations observed in the primary cancer.
A breast cancer study that analyzed 39 matched pairs of primary breast cancers and brain metastases using primer extension mass spectrometry investigated mutations in EGFR, HRAS, KRAS, NRAS, and PIK3CA.
NRAS and PIK3CA mutations were identified independently in samples from two patients, in both the primary and metastatic specimens.
An EGFR mutation was identified in breast cancer from a single patient, but not in the matched metastasis.
From studies of Japanese populations, the frequencies of EGFR mutations in MBTs from non-small cell lung cancer (NSCLC) are similar to those reported for the primary tumor in the same population.
Results from studies investigating EGFR mutations in NSCLC-derived MBTs from Caucasian patients indicate that the mutation frequency in EGFR is less than or equal to 2%, markedly less frequent than the estimated 10% in NSCLC primary tumors.
However, where a mutation was discovered in the metastasis, it was concordant with a mutation in the primary cancer.
These results indicate that coding mutations in the investigated oncogenes are either equivalent or less represented in the MBT compared to the primary tumor, although the body of work is inadequate to draw any conclusions with certainty.
However, evidence suggests that the incidence of mutations in tumor suppressor genes may increase in metastatic cancer.
Whether or not an implicated driver oncogene is discovered in both the primary and metastatic cancer or solely in the brain metastasis is an important distinction that provides rationale for adapting targeted treatments, or developing predictive biomarkers for MBTs.
In a study published in the journal Naturein 2010, Li Ding and co-authors directly investigated whether processes leading to macrometastases are driven by mutations that pre-exist in primary tumor cells or arise at the distant site.
The authors used massively parallel DNA sequencing methods to analyze three related specimens:
- a primary triple negative basal-like breast cancer
- the tumor derived from a mouse xenograft model
- the matched MBT that developed within 8 months of primary diagnosis
For coding mutations, 48 of 50 validated somatic gene mutations were detected in DNA isolated from all three tumor samples, supporting the notion that coding mutations are consistent for primary and metastatic cancer.
The mutation allele frequency for 26 of those was significantly increased in the metastases or xenograft specimens, and the allelic frequency of two mutations was decreased.
The observed levels of enrichment and loss of enrichment (but still detectable) for specific coding mutations in the metastasis and xenograft specimens may indicate that a subset of primary tumor cells carrying the array of mutations in the primary, not just a single clone, were contributing.
Unlike coding mutations, copy number aberrations were gained in the metastasis and xenograft specimens, such that 19% and 39%, respectively, were unique compared to the primary cancer.
Comparable work also using massively parallel sequencing to investigate an estrogen receptor alpha-positive breast tumor arrived at a different conclusion.
Here, the metastasis was detected nine years after the initial diagnosis.
Of the 32 non-synonymous coding mutations detected in the metastatic cancer:
- 19 were absent in the primary tumor
- five were prevalent in the primary tumor
- six were present at lower allelic frequency levels than the primary tumor
The list of mutated genes identified in these studies of individuals has little translational value because a substantial number of specimens are required to elucidate specific, recurring causally implicated gene mutations.
However, these two studies provide highly valuable insights surrounding the potential for sequence and structural mutations and the potential for varied mechanisms driving metastatic cancer associated with short versus long latency.
Somatic Copy Number Variations
The observation made by Li Ding and co-authors that the incidence of copy number alterations is increased in metastasis, and xenograft specimens lend itself to the re-interpretation of other studies.
For example, low-resolution arrayed, comparative genomic hybridization (aCGH) studies have highlighted recurring, broad copy number variation in primary cancers that develop brain metastases, positing that the specific loci represent risk markers of functional consequence.
Further validation is required as to whether the broad regions harbor true driver lesions, or if they represent greater genomic instability underlying other structural mutations with a role in metastasis.
Nevertheless, studies do show significant frequencies of copy number aberrations in specific known cancer genes in brain metastases.
Two studies of breast cancer brain metastasis suggest that gains in EGFR oncogene and loss of PTEN tumor suppressor are more frequent in metastases, one using aCGH methods and the other fluorescent in situ hybridization (FISH).
Two additional studies reporting on FISH analysis of lung cancer brain metastases suggest a marginally higher frequency of EGFR copy number gain (by amplification or polysomy) in brain metastases compared to the matched primary cancer.
Markedly high rates of brain metastases are observed for patients with HER2-amplified breast cancers.
Indications are that the frequency of HER2-positive status is increased in brain metastases over other peripheral metastases, although this may be confounded using HER2-targeted treatments that may be more effective in the treatment of peripheral metastases and restricted in reaching brain tumors.
Additional work by Diane Palmieri and her co-authors in their study published in the journal Cancer Researchin 2007 that analyzed HER2 expression in 124 specimens reinforces that the rate of HER2 overexpression largely associated with gene amplification is 36% in brain metastases.
That is greater than or equal to 10% increase over rates of amplification and overexpression reported in primary breast cancers.
The functional consequence of HER2 amplification and overexpression was demonstrated by this same group using a human cancer cell line model of forced HER2 overexpression, which, when introduced into mice, yielded threefold greater numbers of brain macrometastases compared to the parent cell line xenografts.
Whole-Genome Expression Studies
The variety of possible structural and coding mutations and epigenetic deregulation establish the foundation for aberrant gene expression that is the biomolecular link from the aberrant genome to transformed cancer cell function.
Whole-genome and transcriptome studies of primary cancers are used to identify genes or transcripts displaying significant cancer-specific expression and are especially useful to elucidate cancer cell-specific systems and pathway-level dysfunction.
Additionally, diagnostics based on gene expression signatures have proven effective at predicting lymph node metastases and improving patient outcomes.
Limited recent gene expression data also suggest mechanisms underlying brain metastasis.
In a study published in the journal Breast Cancer Researchin 2010, Leonard Da Silva and his co-authors compared 15 matched primary triple negative, basal breast cancers, and their matched brain metastatic cancers using microarrays that simultaneously analyzed 512 cancer-linked genes.
Twenty-seven genes differed significantly, notably HIF1A and HER3, which were markedly upregulated.
This suggests a role for the epidermal growth factor receptor family in even the triple negative breast cancer subtype.
Also, the HIF1A transcription factor activates genes encoding glucose transporters and glycolytic pathway genes, and an important role for HIF1A is supported by independent work showing more frequent overexpression in patient brain metastases compared to primary breast cancer tissues and enhanced aerobic glycolysis in cells migrating to or implanted in the brain versus parent cells in mouse models of human cancer cell line xenografts.
Multiple lines of whole-genome investigation in cell lines and patient specimens by Paula D. Bos and her co-authors of a study published in the journal Naturein 2009 identified the following as factors contributing to their ability to cross the BBB:
- upregulated expression of an EGFR ligand HBEGF and cyclooxygenase PTGS2
- the ectopic expression of the brain-specific gene α2,6-sialyltransferase ST6GALNAC5 in breast cancer cells
Increased PTGS2 expression is also identified as an important factor in brain metastasizing melanoma.
While the described studies highlight what is unique to metastatic cancer and do not address when or how the features arise, multiple studies present evidence for many, potentially coexisting possibilities.
Studies combining analysis of methylation and gene expression to elucidate recurring features in breast cancer specimens associated with the incidence of brain metastasis do suggest pre-existing features supportive of metastasis, particularly related to genes implicated in EMT.
Analysis of substantial numbers of primary and metastatic breast tumors using whole-genome expression assays was successful in identifying gene expression signatures preserved in both primary and metastatic cancers, suggesting that the bulk of tumor cells in the primary lesion and the metastasis are similar.
Following a xenograft model of metastases, Park Eun Sung and co-authors, in their study published in the journal Proceedings of the National Academy of Sciences of the United States of Americain 2011, present specific evidence that metastatic breast cancer cells acquire expression patterns found in neuronal cells.
Thus, indicating that an evolutionary process underlies MBT development.
Conclusion
The current lack of insight into brain metastases spawns many questions, including whether or not the primary cancer drivers are also the drivers of the related MBT.
Conversely, early data suggests that unique drivers may be responsible for MBT development, raising additional key questions.
Does a new driver/s emerge with the gain of mutations at the metastatic site?
Or alternatively, do cells derived from a minor subclone in the original tumor harbor lesions with the capacity to evade primary adjuvant therapy and seed the metastatic occurrence?
Much research is still needed about molecular genomics of metastatic brain tumors to fill the void and answer these critical questions.

Suleman Shah
Author
Suleman Shah is a researcher and freelance writer. As a researcher, he has worked with MNS University of Agriculture, Multan (Pakistan) and Texas A & M University (USA). He regularly writes science articles and blogs for science news website immersse.com and open access publishers OA Publishing London and Scientific Times. He loves to keep himself updated on scientific developments and convert these developments into everyday language to update the readers about the developments in the scientific era. His primary research focus is Plant sciences, and he contributed to this field by publishing his research in scientific journals and presenting his work at many Conferences.
Shah graduated from the University of Agriculture Faisalabad (Pakistan) and started his professional carrier with Jaffer Agro Services and later with the Agriculture Department of the Government of Pakistan. His research interest compelled and attracted him to proceed with his carrier in Plant sciences research. So, he started his Ph.D. in Soil Science at MNS University of Agriculture Multan (Pakistan). Later, he started working as a visiting scholar with Texas A&M University (USA).
Shah’s experience with big Open Excess publishers like Springers, Frontiers, MDPI, etc., testified to his belief in Open Access as a barrier-removing mechanism between researchers and the readers of their research. Shah believes that Open Access is revolutionizing the publication process and benefitting research in all fields.

Han Ju
Reviewer
Hello! I'm Han Ju, the heart behind World Wide Journals. My life is a unique tapestry woven from the threads of news, spirituality, and science, enriched by melodies from my guitar. Raised amidst tales of the ancient and the arcane, I developed a keen eye for the stories that truly matter. Through my work, I seek to bridge the seen with the unseen, marrying the rigor of science with the depth of spirituality.
Each article at World Wide Journals is a piece of this ongoing quest, blending analysis with personal reflection. Whether exploring quantum frontiers or strumming chords under the stars, my aim is to inspire and provoke thought, inviting you into a world where every discovery is a note in the grand symphony of existence.
Welcome aboard this journey of insight and exploration, where curiosity leads and music guides.
Latest Articles
Popular Articles